Не так давно команда разработчиков сервиса Blogger запустила площадку Blogger In Draft, на которой в открытом для всех зарегистрированных пользователей режиме тестируются новые функции этого блог-хостинга.
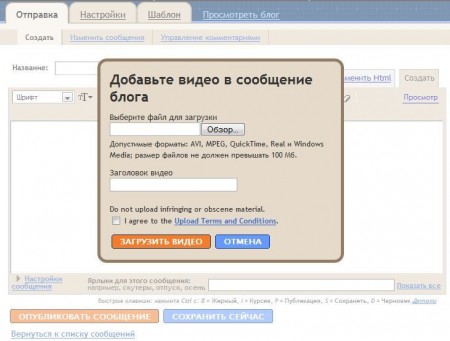
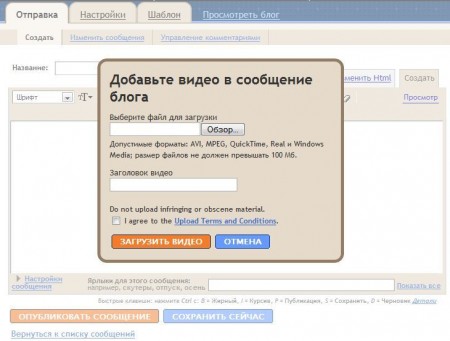
Первой такой функцией стало размещение видео в постах. Правда, имеется в виду не интеграция с YouTube, как было бы более логично, а закачка файла с локального компьютера. При размещении видео нужно согласиться с условиями предоставления услуги: ссылка на них ведёт на сайт Google Video, который хоть и становится видео-поисковиком, но продолжает принимать ролики от пользователей. Пример размещённого видео можно увидеть здесь.
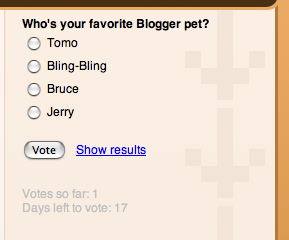
Вчера на Blogger In Draft появилась новая опция — голосования, которые пользователь может поместить в боковой колонке своего блога.

Кроме того, теперь в RSS-потоки своего блога можно встроить ссылки на медиа-файлы, чтобы пользователи iTunes и других программ, использующихся для прослушивания или просмотра подкастов, могли подписываться на RSS блога и закачивать подкасты на свой компьютер или плейер.
Естественно, все эти возможности доступны пока только через интерфейс Blogger In Draft.
Первой такой функцией стало размещение видео в постах. Правда, имеется в виду не интеграция с YouTube, как было бы более логично, а закачка файла с локального компьютера. При размещении видео нужно согласиться с условиями предоставления услуги: ссылка на них ведёт на сайт Google Video, который хоть и становится видео-поисковиком, но продолжает принимать ролики от пользователей. Пример размещённого видео можно увидеть здесь.
Вчера на Blogger In Draft появилась новая опция — голосования, которые пользователь может поместить в боковой колонке своего блога.
Кроме того, теперь в RSS-потоки своего блога можно встроить ссылки на медиа-файлы, чтобы пользователи iTunes и других программ, использующихся для прослушивания или просмотра подкастов, могли подписываться на RSS блога и закачивать подкасты на свой компьютер или плейер.
Естественно, все эти возможности доступны пока только через интерфейс Blogger In Draft.













